Транскрипция,
ее регуляция,
и как ее изучают
(с помощью секвенирования и биоинформатики)
Ваня Кулаковский, Пущино-Москва
Часть 2: анализ ДНК-белкового узнавания in vivo
с помощью высокопроизводительных экспериментальных методов
От связывания факторов транскрипции
к системной биологии


Как фактор транскрипции находит свой геномный адрес?
Как нам выяснить его геномный адрес?
Догеномные методы: EMSA, "гель-шифт"

Watson, Molecular Biology of the Gene, 2014
Догеномные методы: ДНКазный футпринтинг

Watson, Molecular Biology of the Gene, 2014
SELEX (Systematic Evolution of Ligands by Exponential Enrichment)

Watson, Molecular Biology of the Gene, 2014
SELEX (Systematic Evolution of Ligands by Exponential Enrichment)
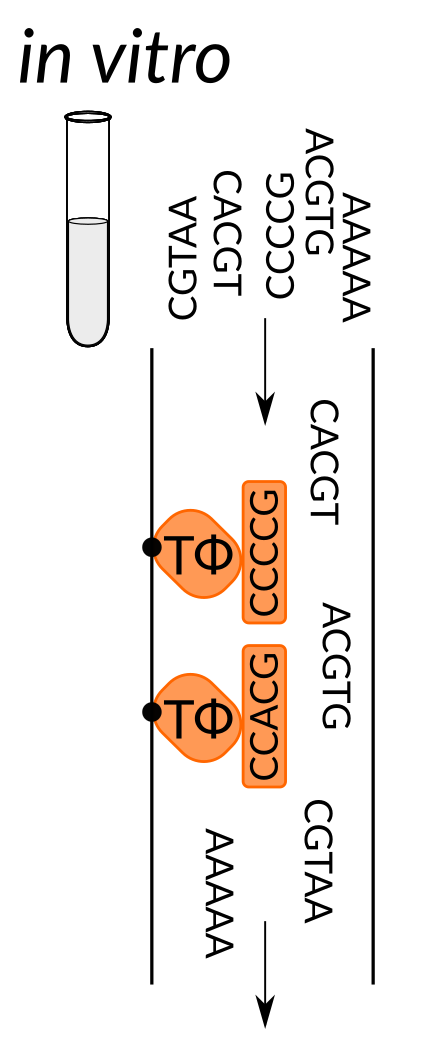
Прямолинейное применение высокопроизводительного секвенирования: полногеномный футпринтинг - профилирование открытого хроматина (DNase-seq), и высокопроизводительный SELEX (HT-SELEX, SELEX-Seq).
ДНК-микрочипы для анализа экспрессии генов
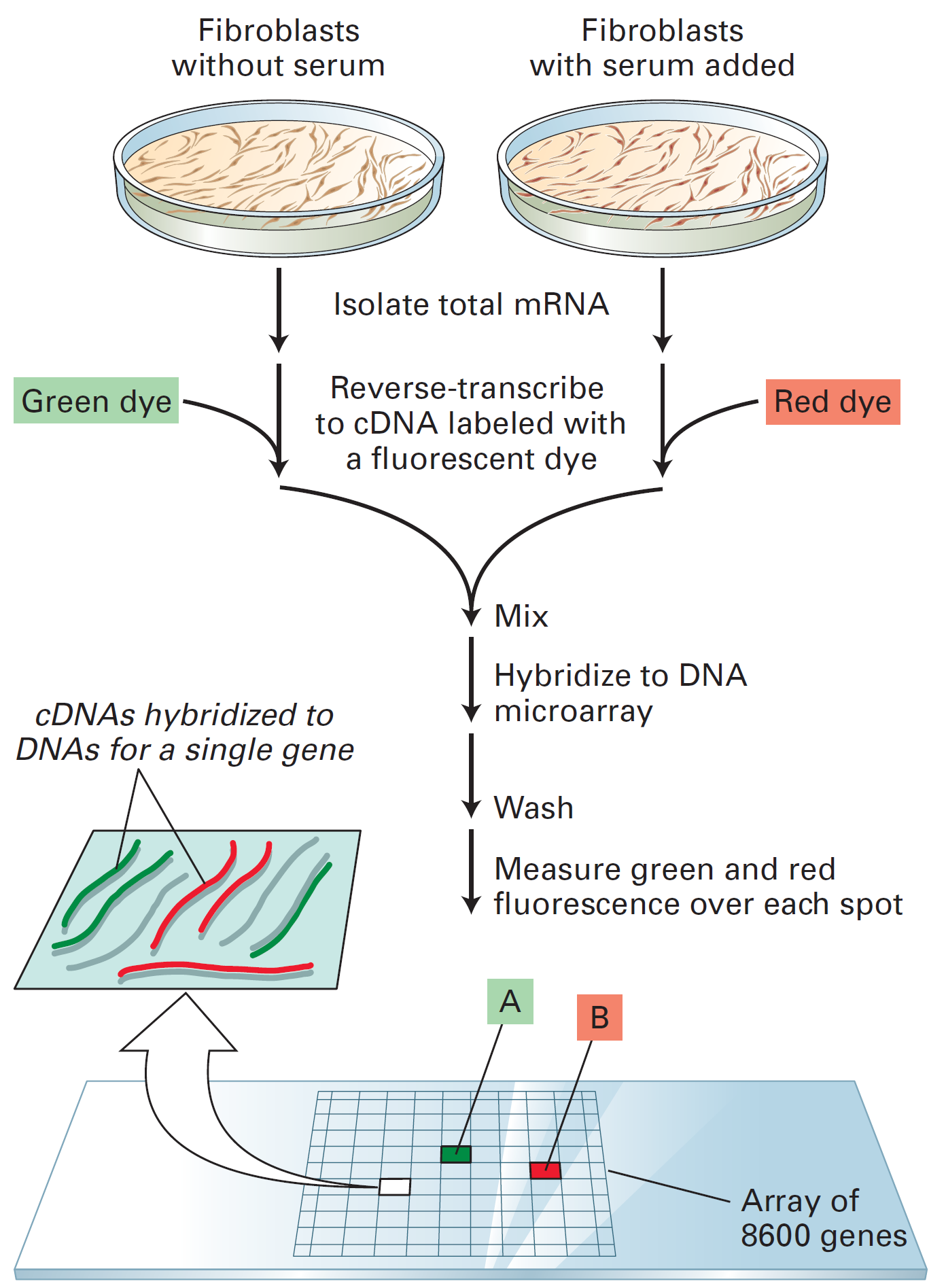
Lodish et al., Molecular Cell Biology, 8th edition, 2016
Первые "большие" методы: гибридизация на микрочипах
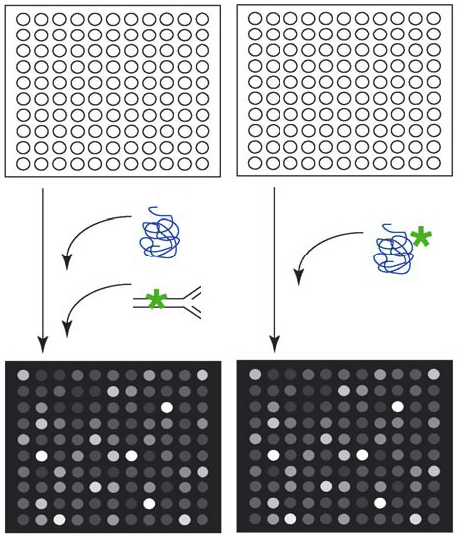
Protein-binding microarray (PBM)
Bulyk 2006, http://www.ncbi.nlm.nih.gov/pubmed/16839757Первые "большие" методы: гибридизация на микрочипах
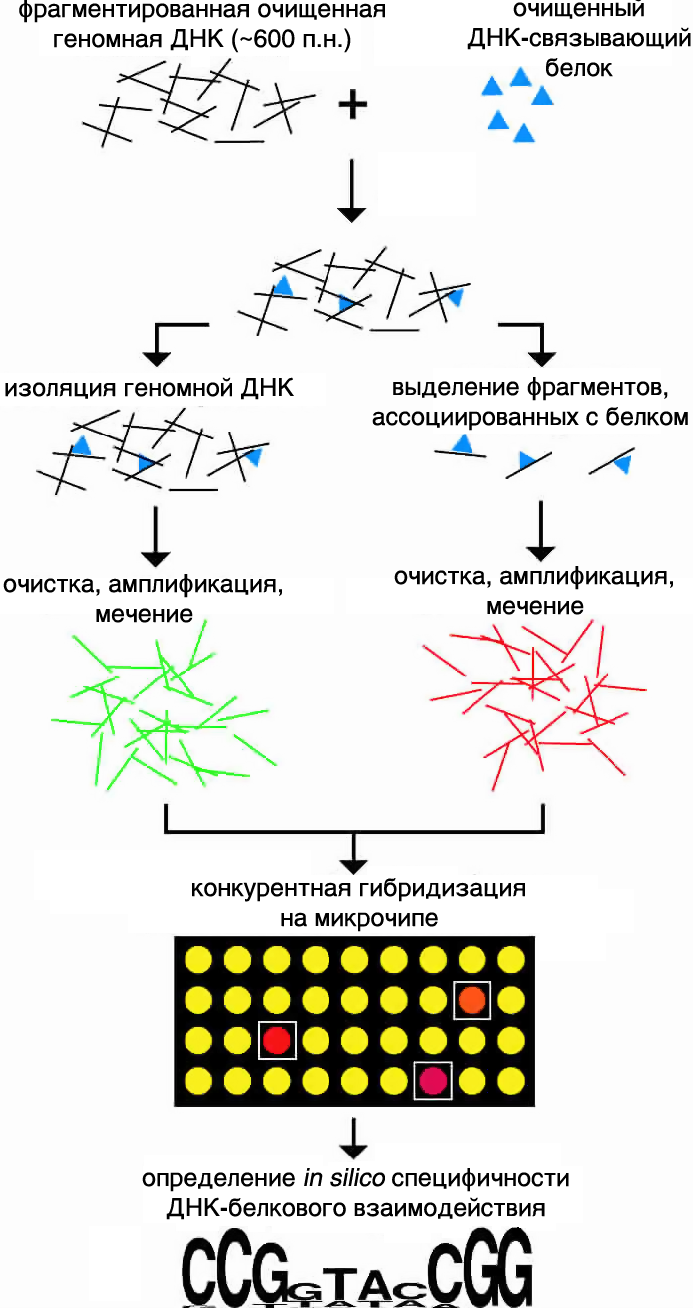
DNA-immunoprecipitation chip (DIP-chip)
Liu 2005, http://www.ncbi.nlm.nih.gov/pubmed/15710749ChIP-on-chip: теперь банановый in vivo
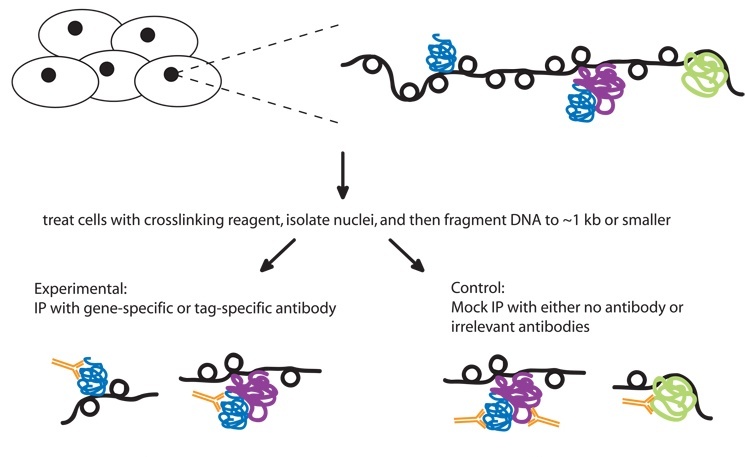
Bulyk 2006, http://www.ncbi.nlm.nih.gov/pubmed/16839757
ChIP-on-chip
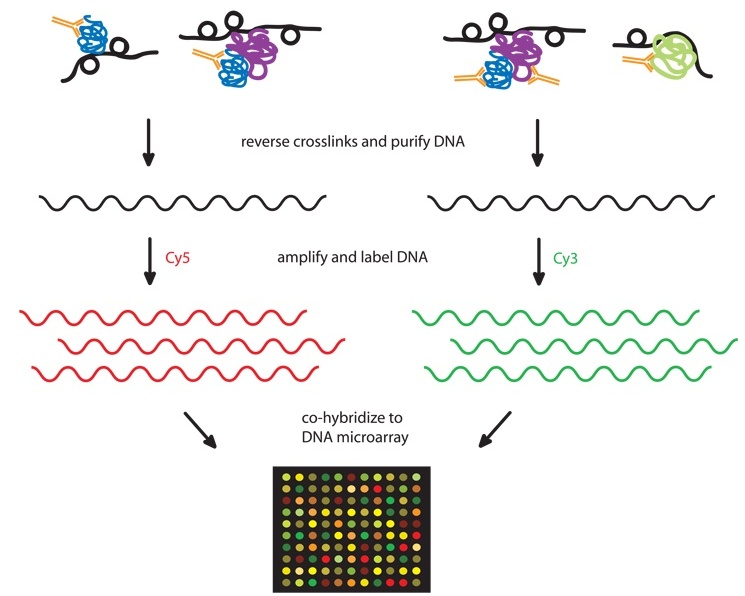
Bulyk 2006, http://www.ncbi.nlm.nih.gov/pubmed/16839757

Интернеты
ChIP-Seq: chromatin immunoprecipitation
followed by deep sequencing
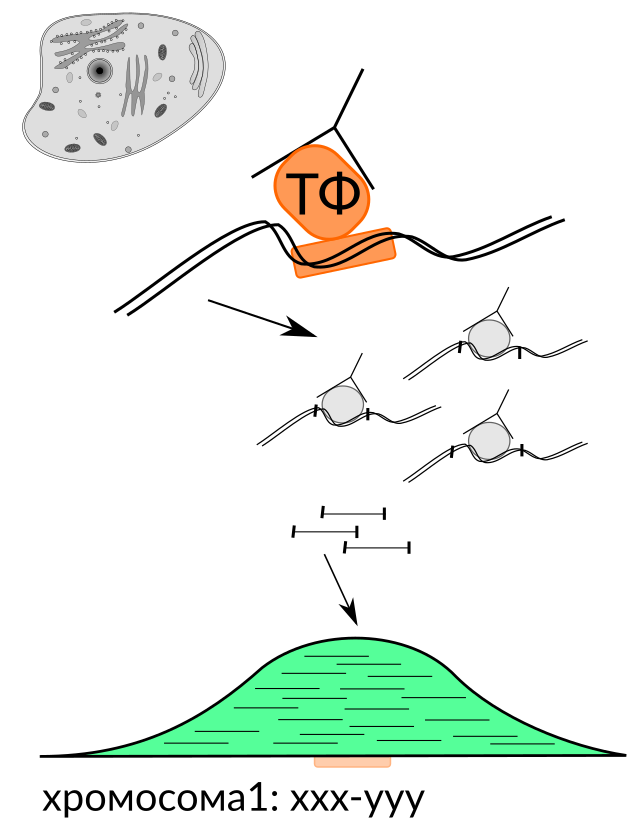
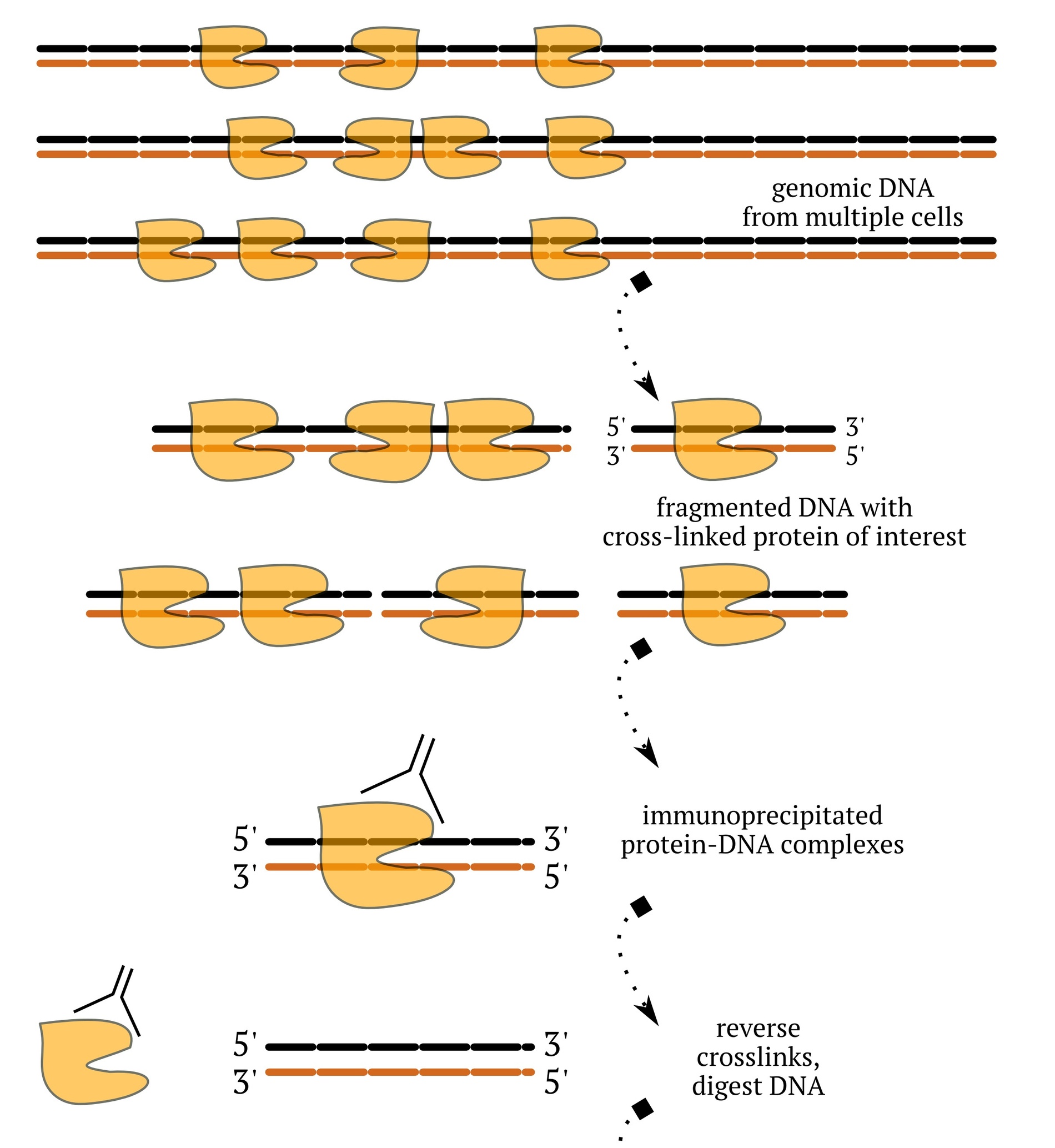
Kulakovskiy and Makeev, 2013, http://www.ncbi.nlm.nih.gov/pubmed/23790213
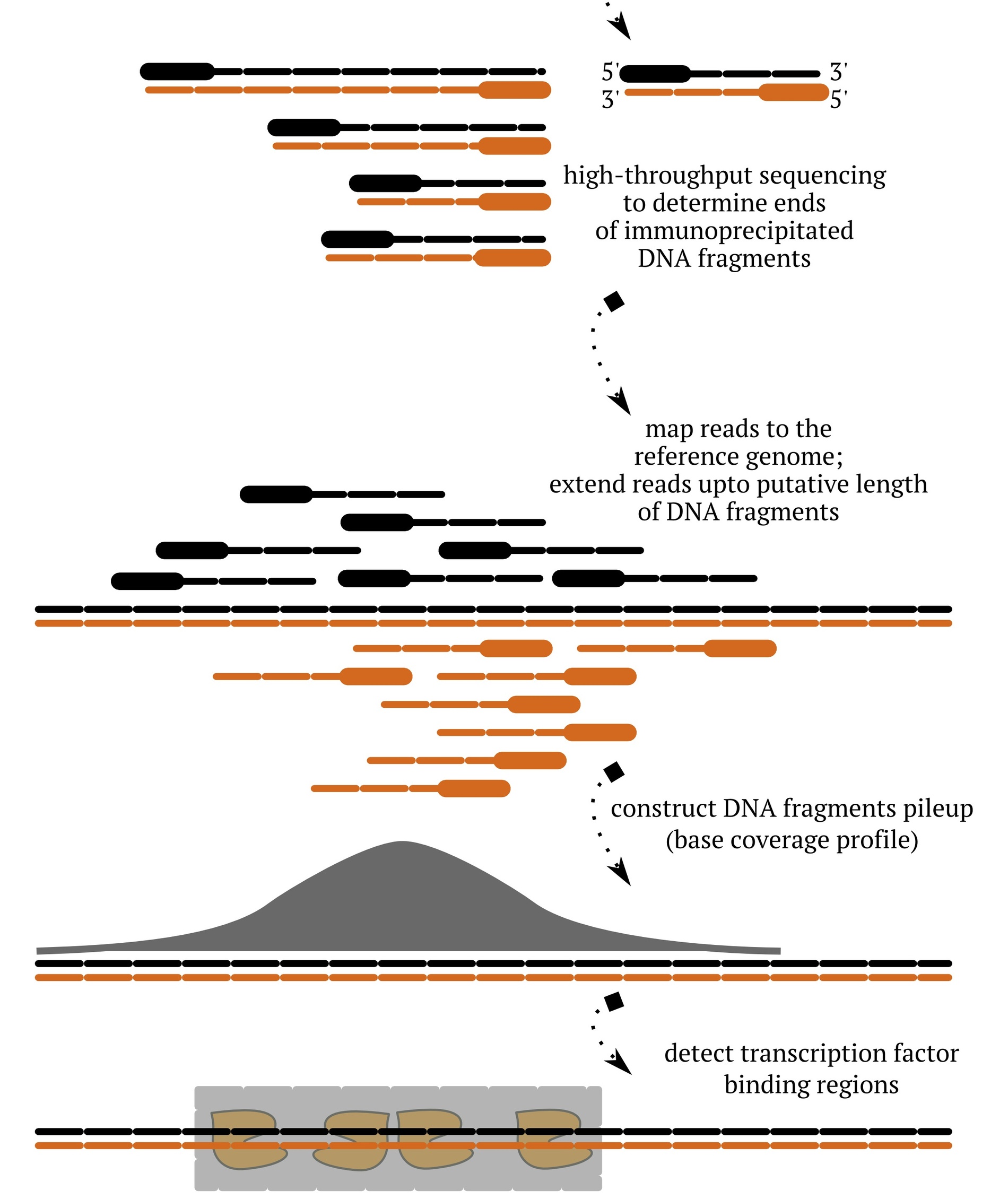
Kulakovskiy and Makeev, 2013, http://www.ncbi.nlm.nih.gov/pubmed/23790213
ChIP-exo: как ChIP-Seq только лучше
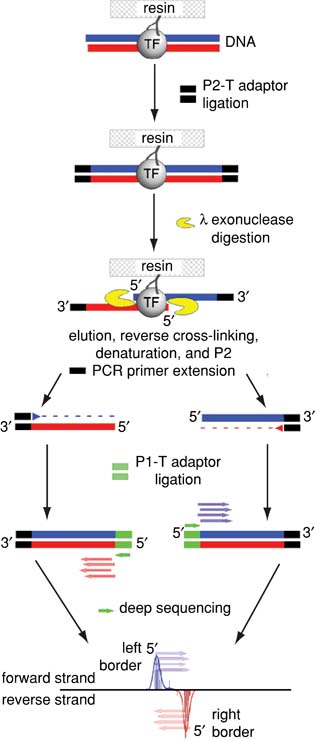
Rhee and Pugh, 2012, http://www.ncbi.nlm.nih.gov/pubmed/23026909
ChIP-exo: гораздо лучше (но есть подвох)
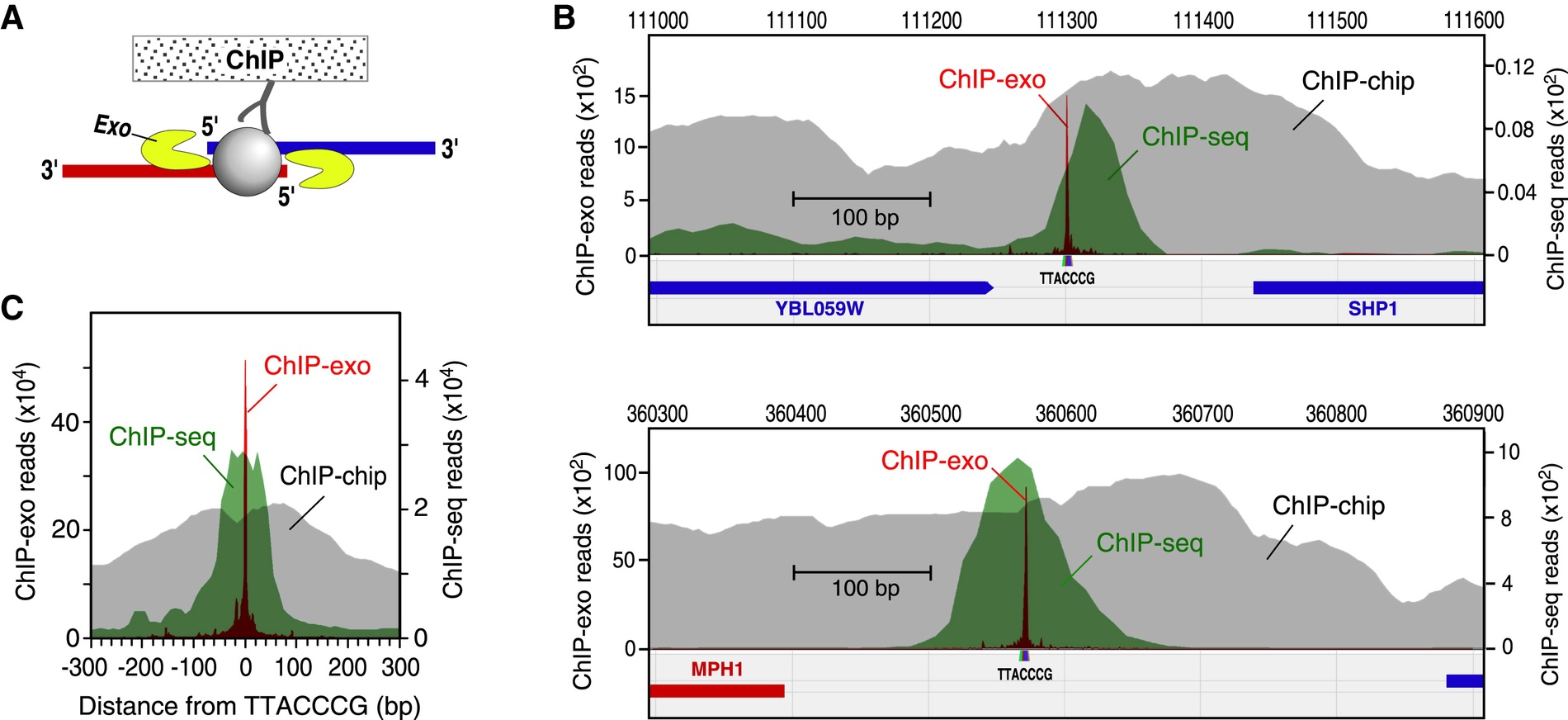
Rhee and Pugh, 2011, http://www.ncbi.nlm.nih.gov/pubmed/22153082
Cut & Run: еще лучше (и снова подвох)
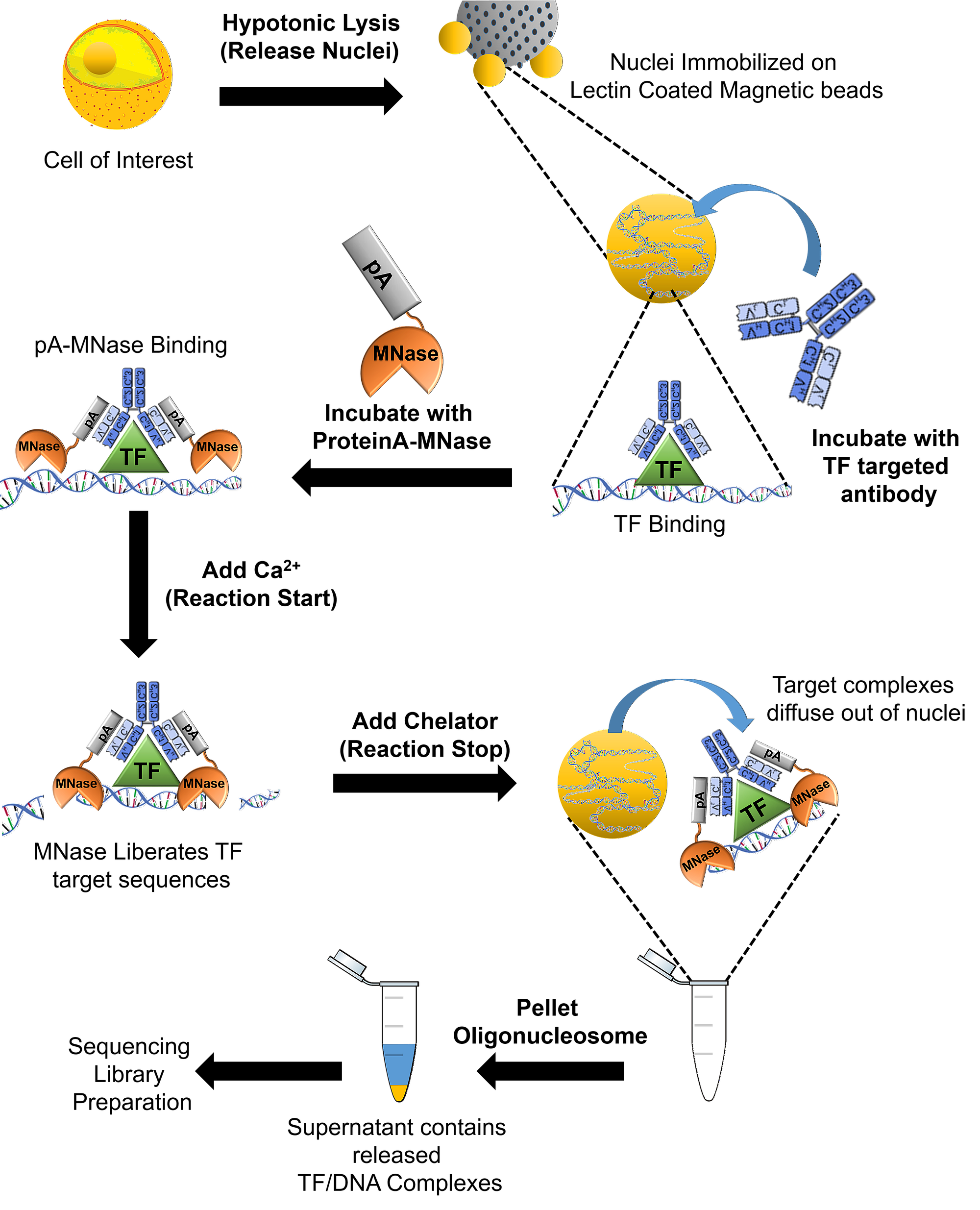
Skene & Henikoff, eLife, 2017; Figure by Mannan369, used under CC BY-SA 4.0
"Several ChIP-free strategies have been developed for scChIP-seq."
Можно использовать для отдельных клеток:
scChIC-(immuno-cleavage)-seq [doi:10.1038/s41592-019-0361-7],
uliCUT&RUN [doi:10.1016/j.cell.2019.03.014] - в некоторых случаях достаточно всего 50 клеток (!)
Для анализа эпигеномов: CUT&Tag [doi:10.1038/s41467-019-09982-5] и аналоги на основе тагментации транспозазой Tn5.
Nakato & Sakata, Methods for ChIP-seq analysis: A practical workflow and advanced applications, Methods, 2020Анализ данных ChIP-Seq
Контрольный эксперимент ChIP-Seq может быть
традиционный: секвенирование геномной ДНК (Input DNA) либо результатов неспецифической иммунопреципитации (нерелевантный белок, без антител; Mock IP),
альтернативный: ChIP-Seq при нокауте или нокдауне целевого белка,
оптимально спуленный: WACS (Awdeh et al., 2019, bioRxiv),
виртуальный: AIControl (Hiranuma et al., 2019, NAR).
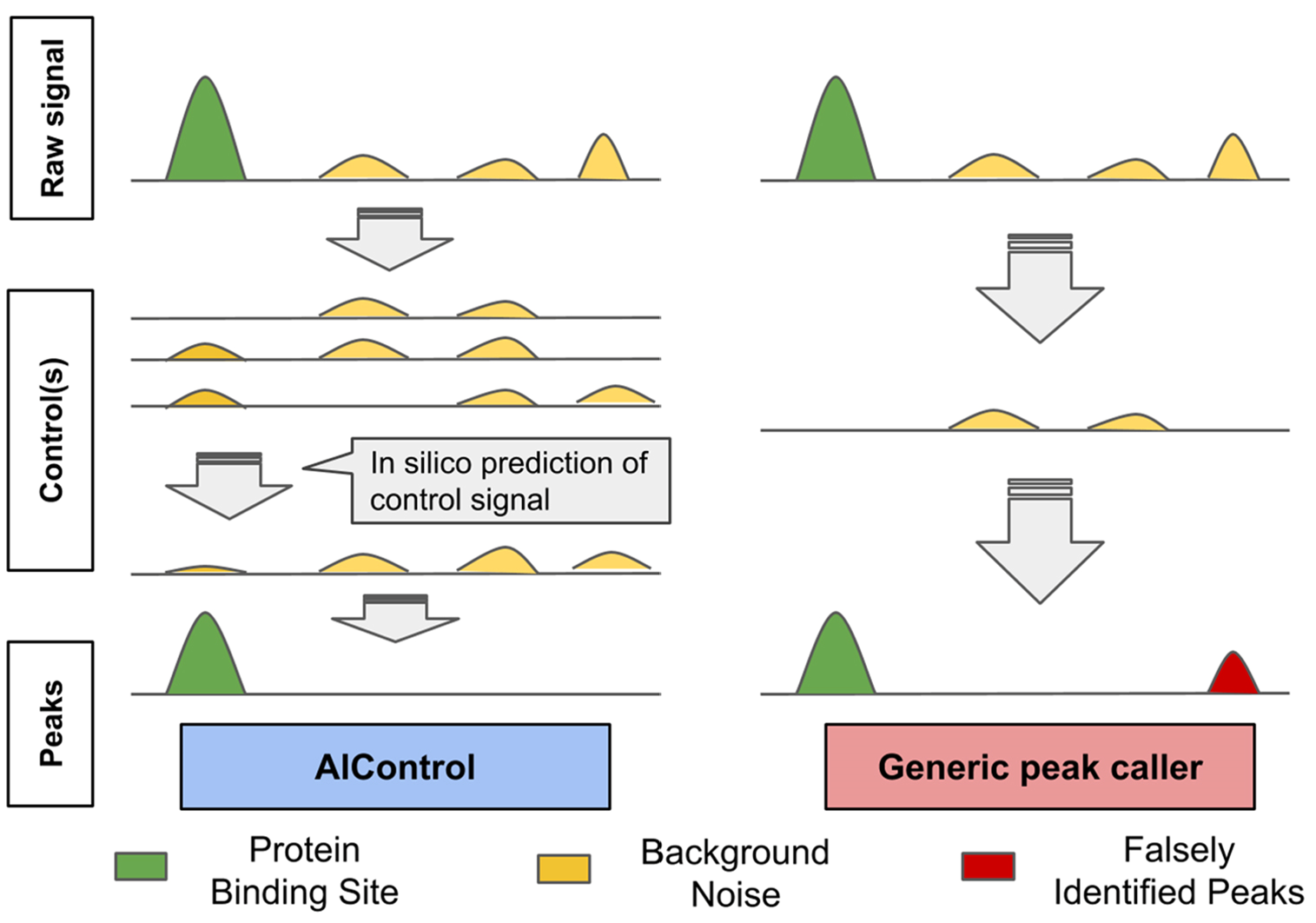
A еще его может и не быть!
Базовая обработка и картирование прочтений
Димеры адаптерных последовательностей
Чаще используются короткие одиночные прочтения (проблема картируемости)
Прочтения (риды) это не сайты связывания и не связываемые фрагменты
Что делать с повторами?
The read mappers are designed to allow a (user-settable) number of mismatches in the reads, and it is important to choose this parameter to be appropriate with the NGS platform being used (consult the manufacturer).T. Bailey in Practical guidelines for the comprehensive analysis of ChIP-seq data, 2014.
Yet-another-Bioinformatics-Hell


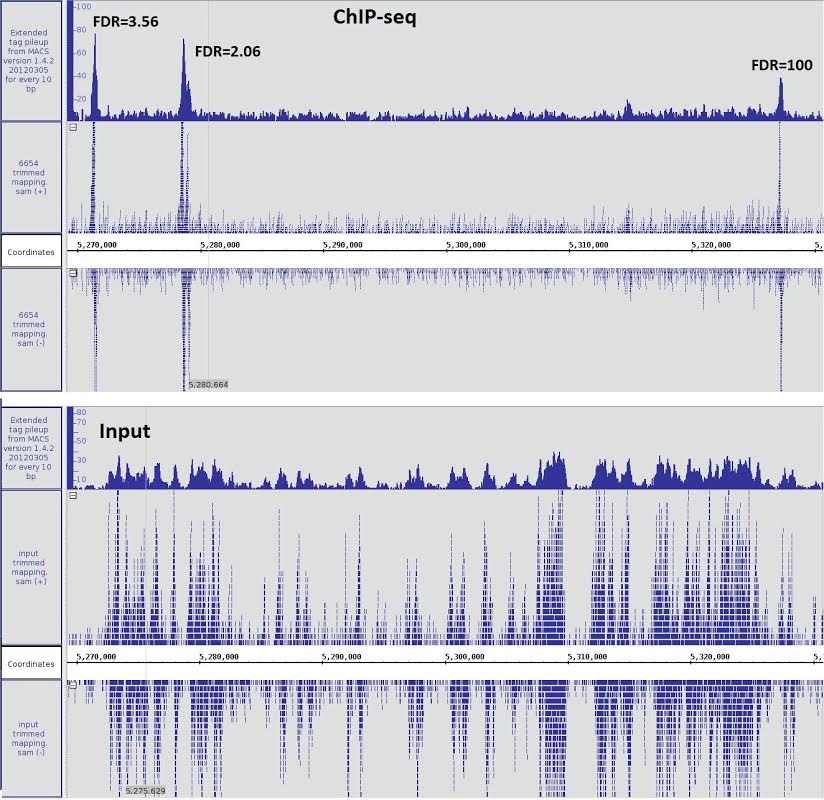
Какой же программой искать пики?
Известные программы эпохи ранней популярности ChIP-Seq:
CisGenome не обновлялся с 2011;
FindPeaks не обновлялся, wiki с документацией недоступно более 5 лет;
SISSRs не обновлялся с 2008 (но все еще работает).
MACS: живой мамонт

Общая схема
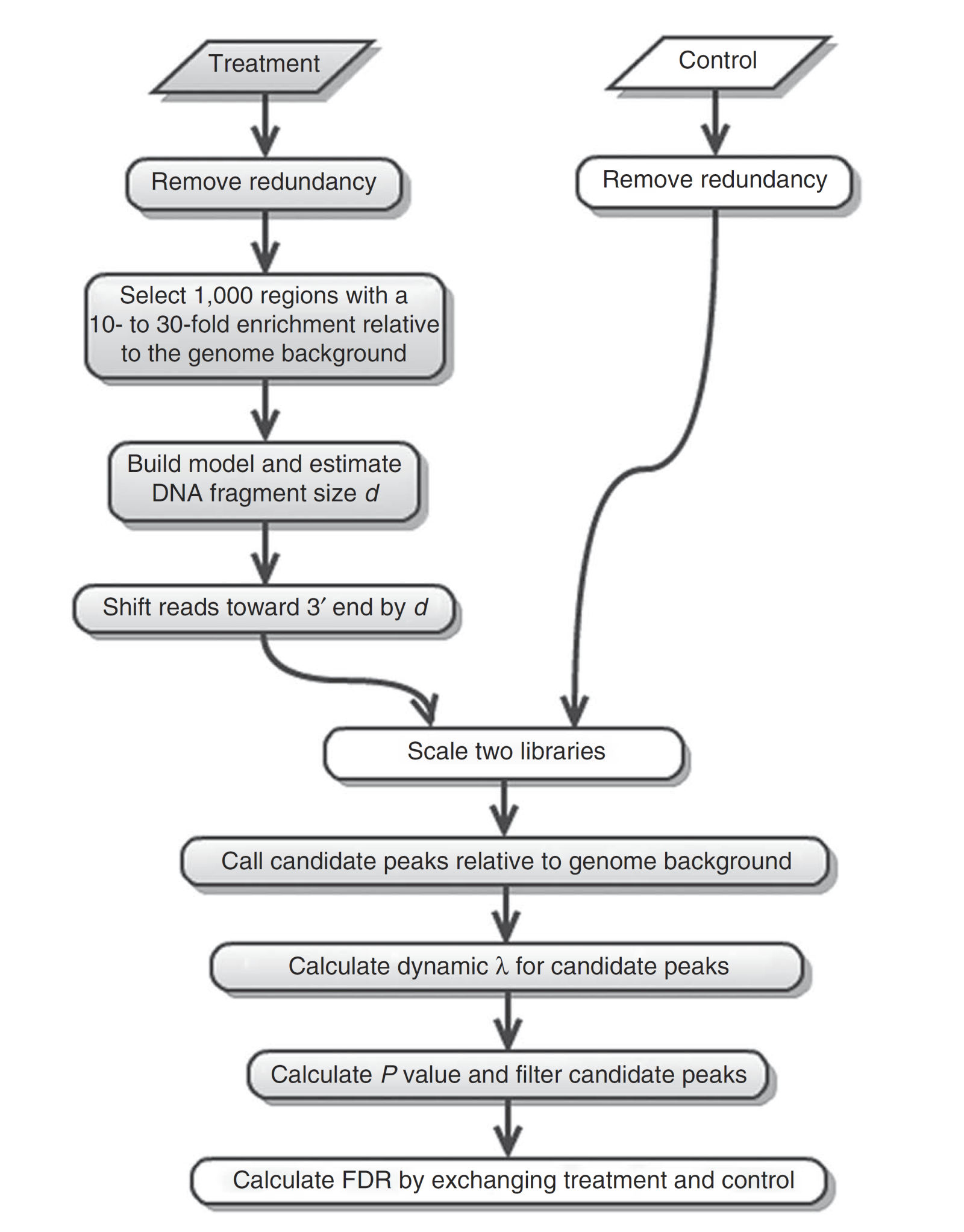
Feng et al. 2012, Nat Protocols
Эмпирическая оценка длин фрагментов ДНК
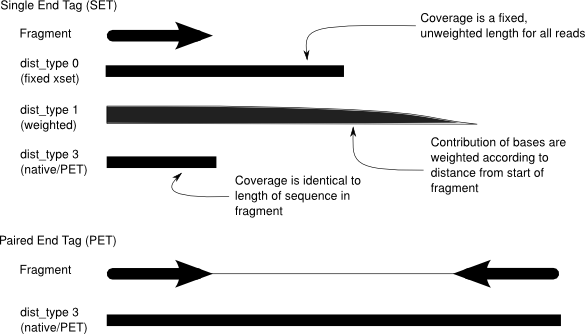
http://sourceforge.net/apps/mediawiki/vancouvershortr/index.php?title=FP4Parameters
Вспомним как получаются пики
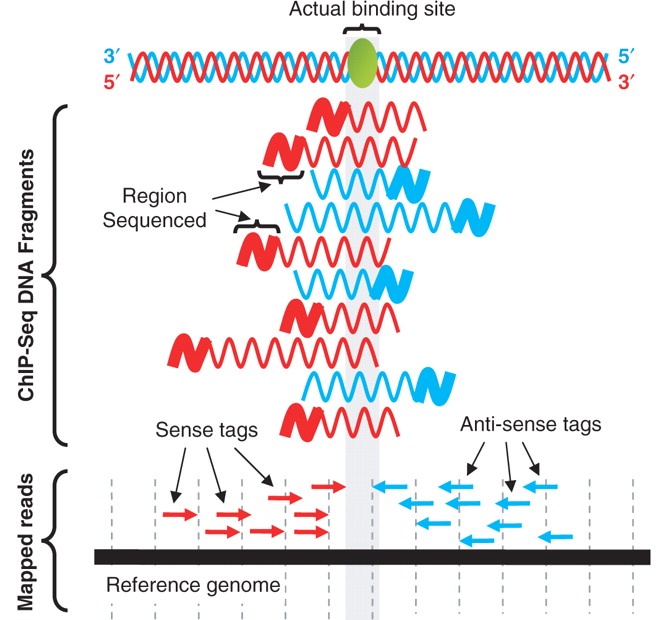
Jothi 2008, http://www.ncbi.nlm.nih.gov/pubmed/18684996
И действительно относительный сдвиг прочтений видно невооруженным взглядом

MACS определяет характерную длину d "полезного" фрагмента и сдвигает прочтения в 3' направлении на d/2
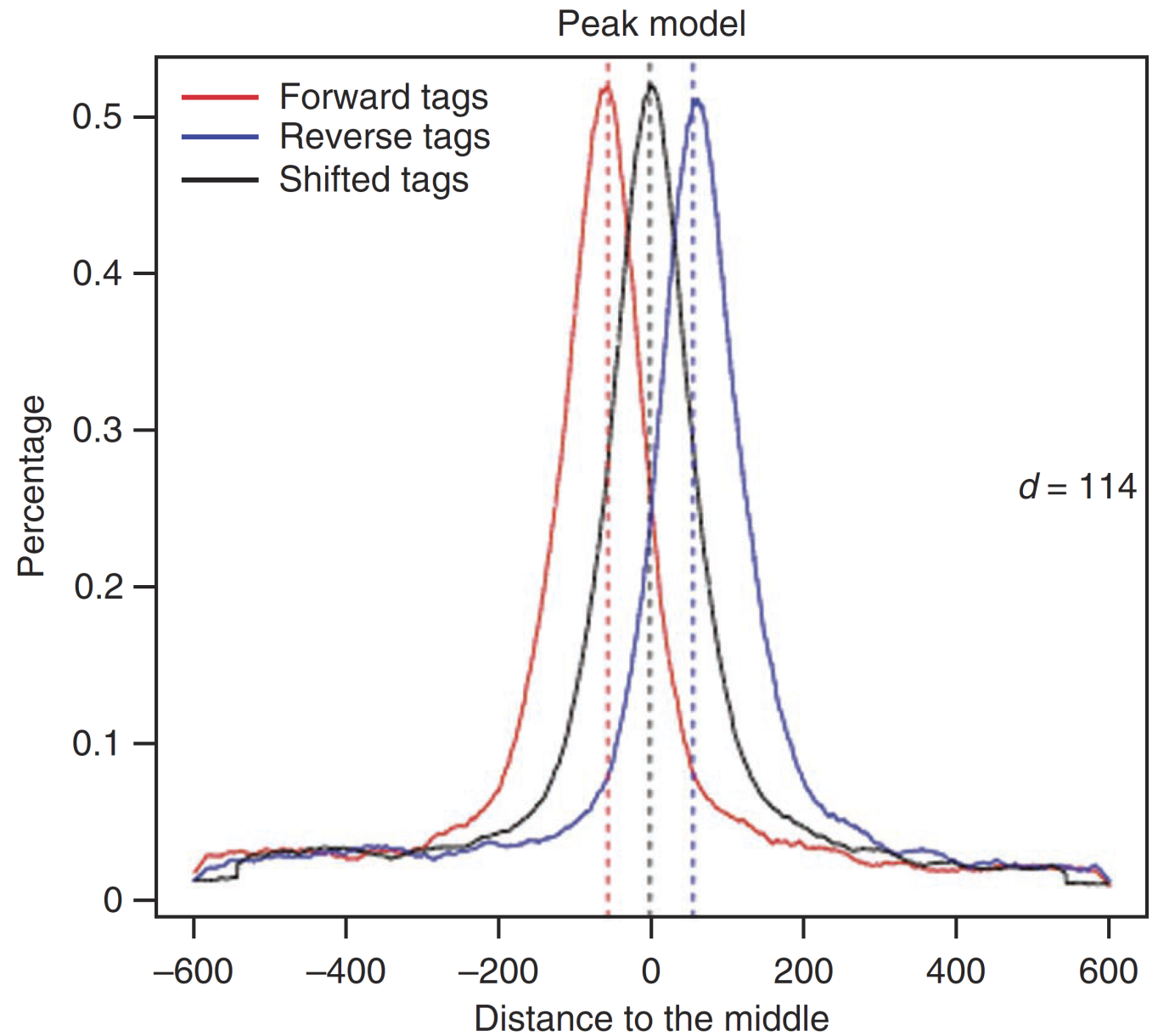
Feng et al. 2012, Nat Protocols
Ожидаемая и наблюдаемая длина фрагмента после соникации
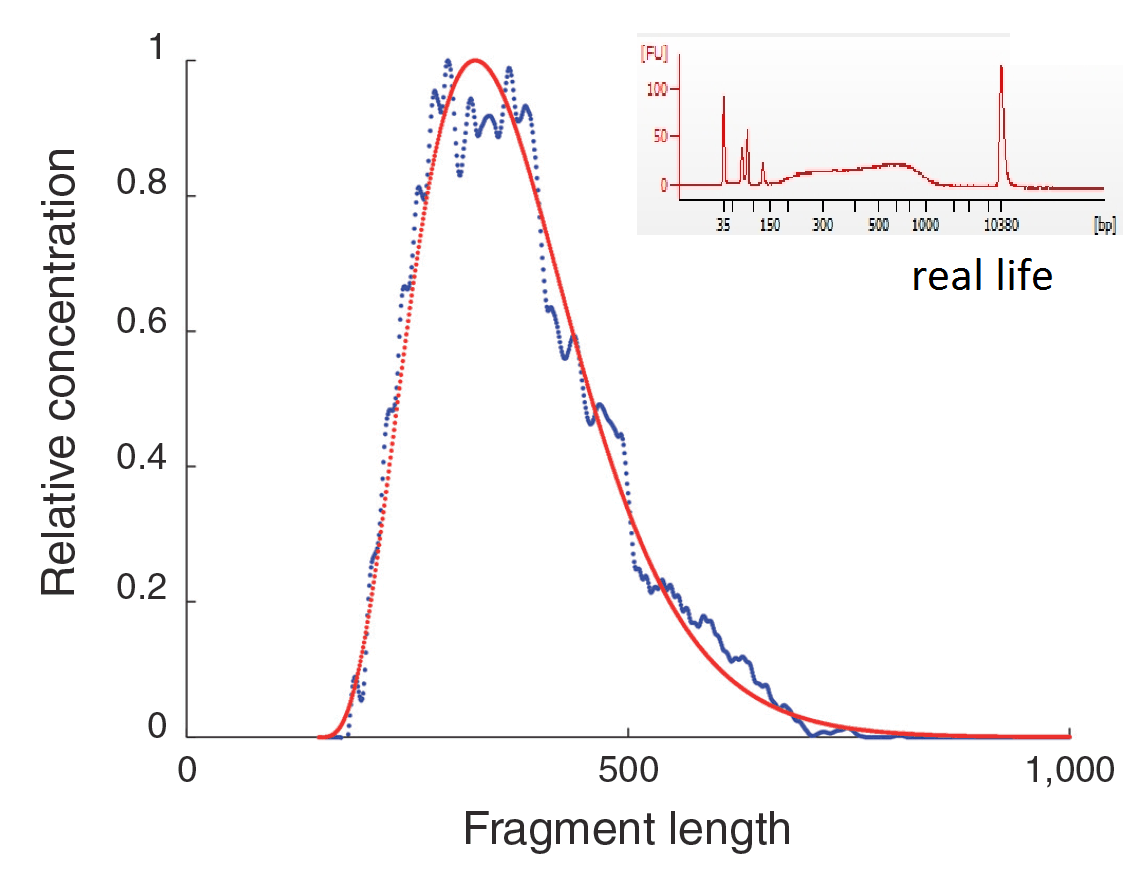
Qi 2006, http://www.ncbi.nlm.nih.gov/pubmed/16900145
Число прочтений моделируется распределением Пуассона (где λ - ожидаемое число прочтений в окне)
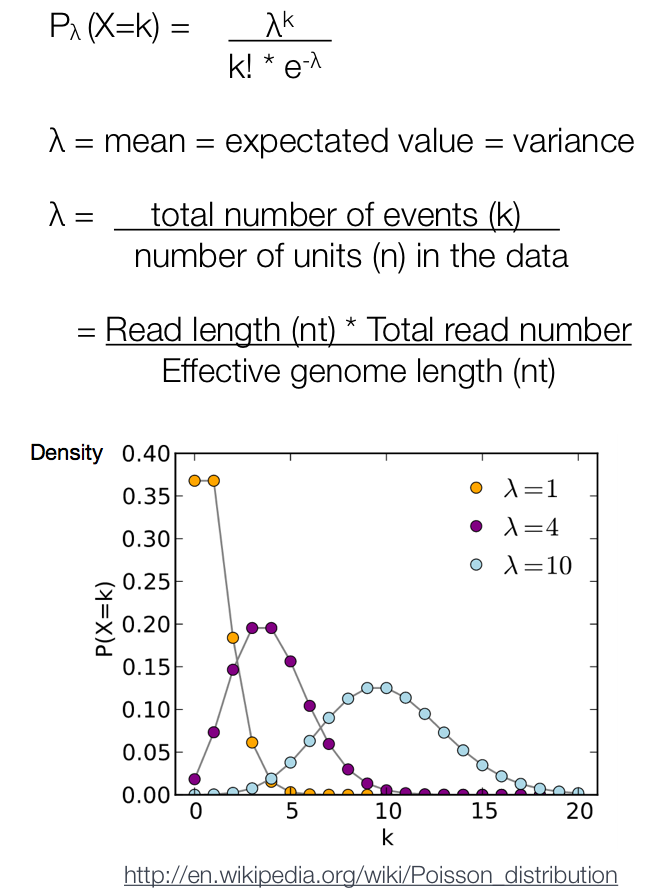
https://hbctraining.github.io/Intro-to-ChIPseq/lessons/05_peak_calling_macs.html
Но бывают некоторые сложности
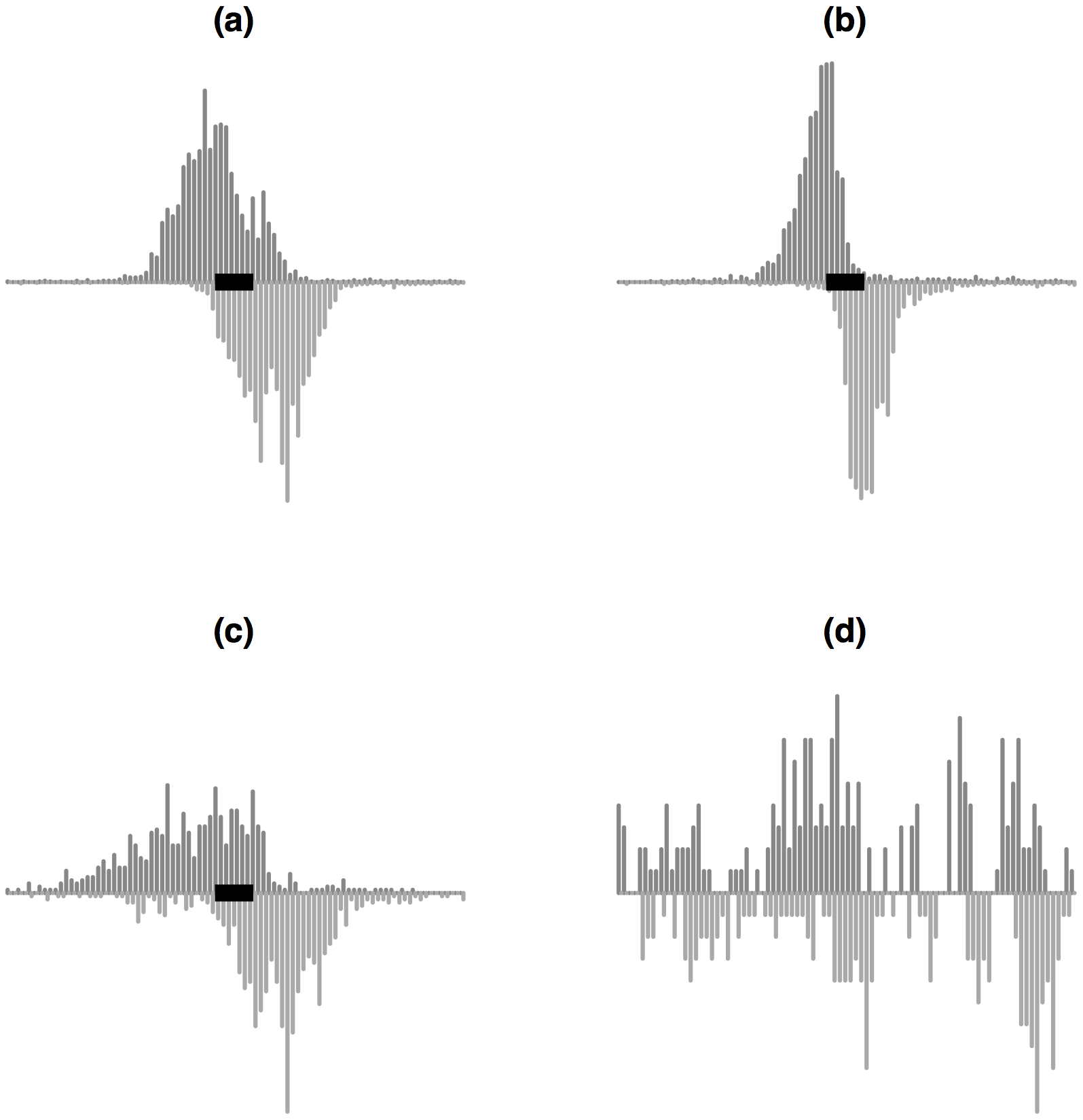
Wu and Ji, 2014, http://www.ncbi.nlm.nih.gov/pubmed/24608116

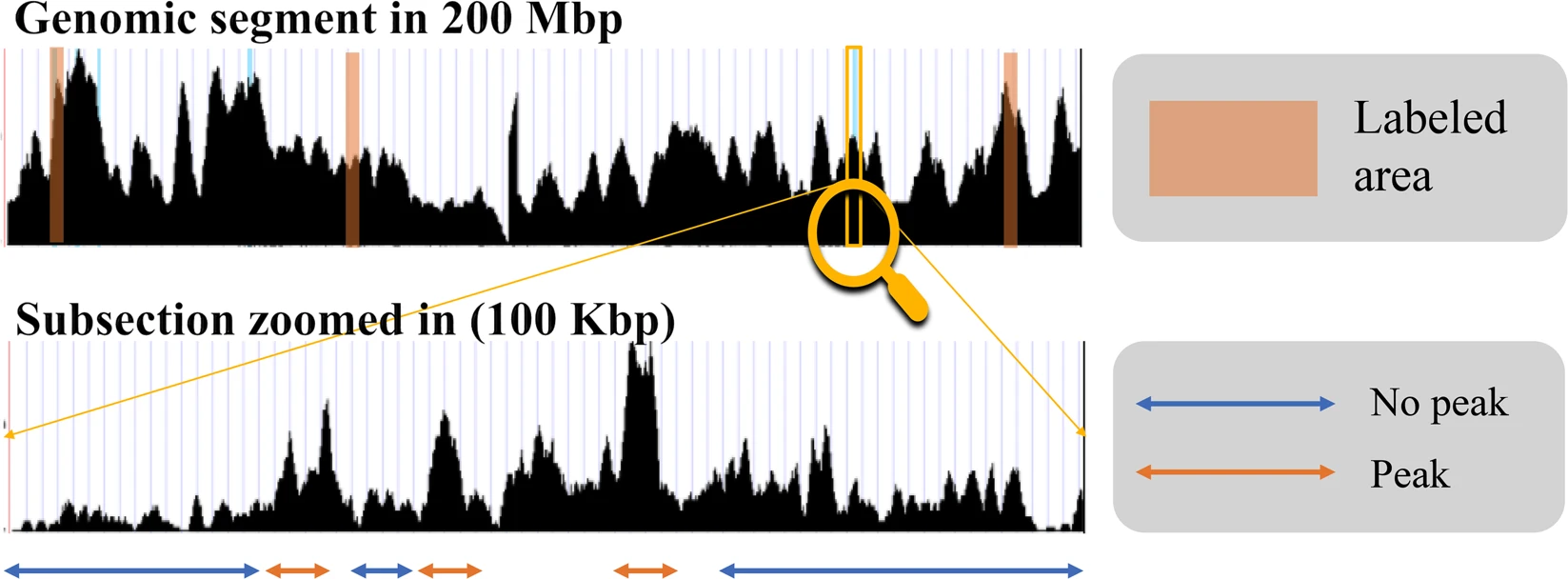

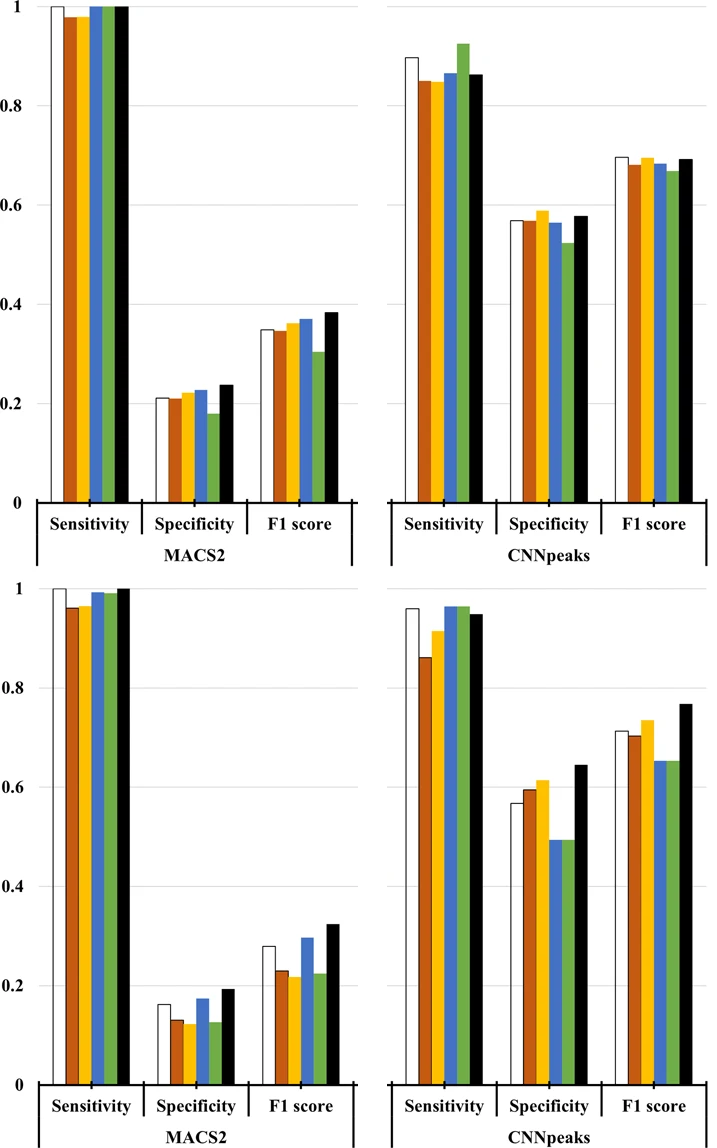
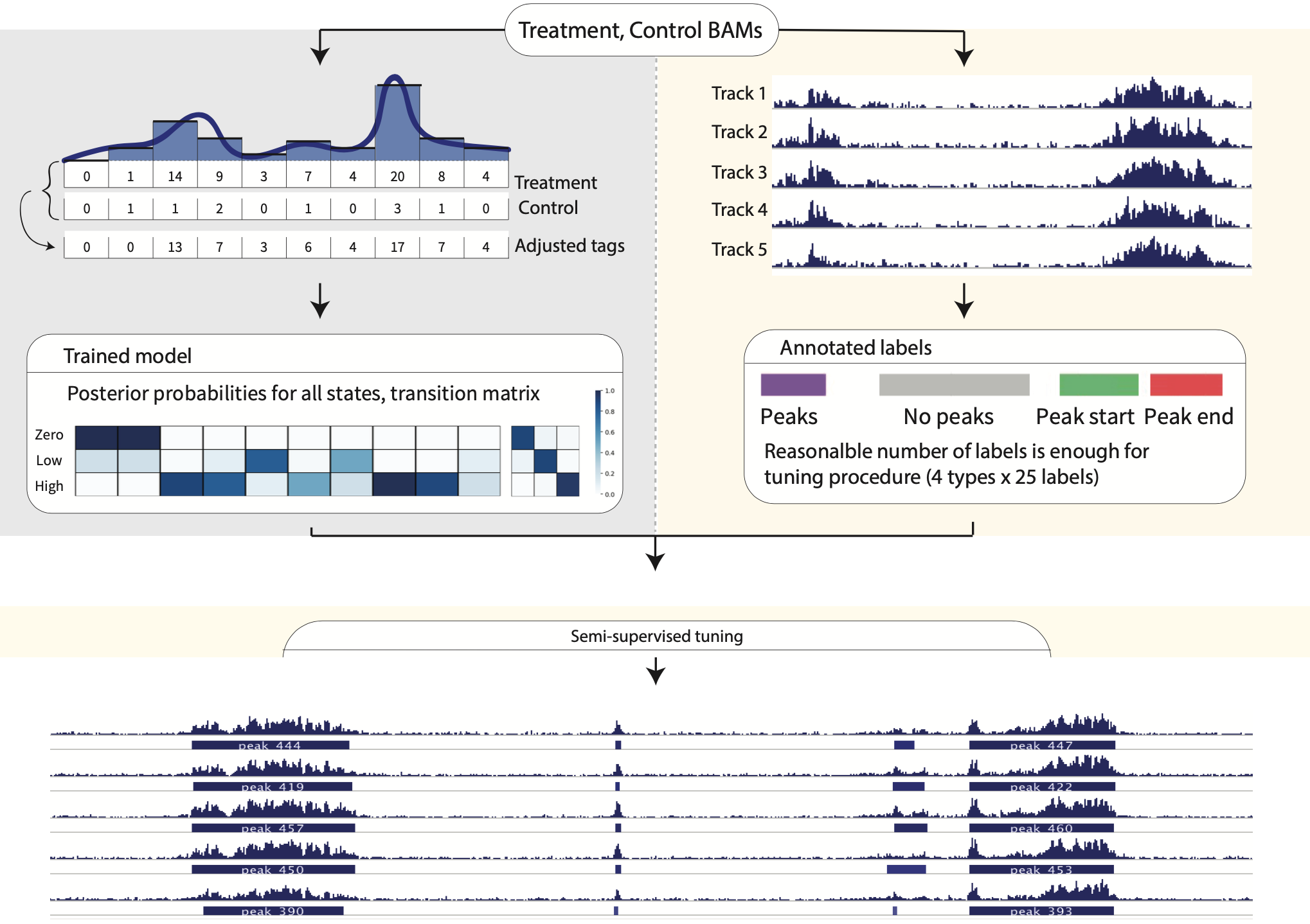
Вручную разметили более 3 тысяч (!) геномных сегментов.
JetBrains Research SPAN
Чипсики - все ли так радужно?

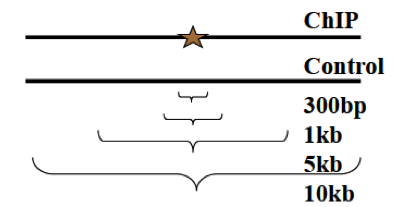
Где эксперимент (IP) и где контроль?
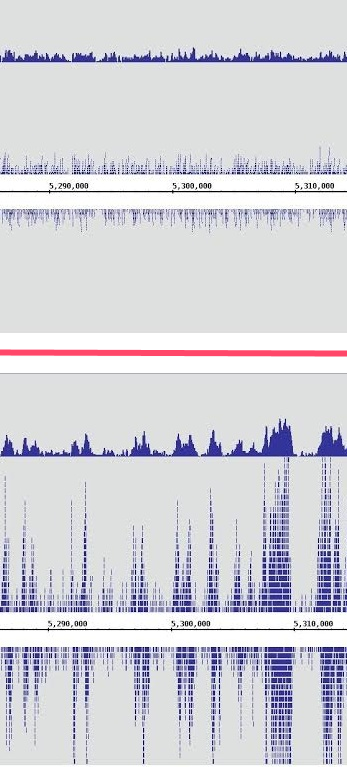
Nickolay Zolotarev, Oksana Maksimenko
Все наоборот
Nickolay Zolotarev, Oksana Maksimenko
Шеф, у нас проблемы

Large-scale quality analysis of published ChIP-seq data, 2014
It is, for example, well appreciated that differential chromatin shearing efficiency can lead to the overrepresentation of areas of open chromatin (usually immediately surrounding transcribed promoters) in sequencing libraries. This has been termed the "Sono-seq" effect when attributed to sonication (Auerbach et al. 2009, Mapping accessible chromatin regions using Sono-Seq).Marinov, 2014, http://www.ncbi.nlm.nih.gov/pubmed/24347632
Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins, 2013
The localization of unrelated proteins, including the entire silencing complex, to the most highly transcribed genes was highly suggestive of a technical issue with the immunoprecipitations.Teytelman, 2013, http://www.ncbi.nlm.nih.gov/pubmed/24173036
Widespread Misinterpretable ChIP-seq Bias in Yeast, 2013
Another type of background bias stems from the inherent nucleosomal structure of chromatin, and can potentially make it seem like certain factors bind nucleosomes even when they don't.Park, 2013, http://www.ncbi.nlm.nih.gov/pubmed/24173036
Large-scale quality analysis of published ChIP-seq data, 2014
Data quality varied widely across "impact" levels. We separated datasets into groups according to the 2011 Thomson Reuters Impact Factor for the journal in which the corresponding article was published, and examined the distribution of QC scores in each group (Supplementary Figure 8). The group with highest impact factor (≥25) contained the largest fraction of datasets with a low QC { SPP in Kharchenko, 2008 } score of -2 or -1.Marinov, 2014, http://www.ncbi.nlm.nih.gov/pubmed/24347632
Using CisGenome to analyze ChIP-chip and ChIP-seq data, 2011
Users should look at the FDR and construct a final peak list using a FDR cutoff they are comfortable with (usually FDR = 10% or less). ...FDR below 25% is usually acceptable.Ji, 2011, http://www.ncbi.nlm.nih.gov/pubmed/21400695

...enrichment in a given ChIP experiment depends on many intractable parameters, likely including a phase of a moon.
Чипсеки вредны?
Полезны, но при интерпретации нужно учитывать: глубину секвенирования, адекватность контроля, вычислительный пайплайн, дополнительные данные (например, анализ мотивов), воспроизводимость.
Что нам поможет? FastQC, ChIPQC https://hbctraining.github.io/Intro-to-ChIPseq/lessons/06_combine_chipQC_and_metrics.html
Черные списки http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/
Рекомендации и стандарты проекта ENCODE




Мера невоспроизводимости
IDR (Irreproducible Discovery Rate)
 Li et al, Measuring reproducibility of high-throughput experiments, Ann. Appl. Stat., 2011
Li et al, Measuring reproducibility of high-throughput experiments, Ann. Appl. Stat., 2011
Каков же геномный адрес факторов транскрипции?
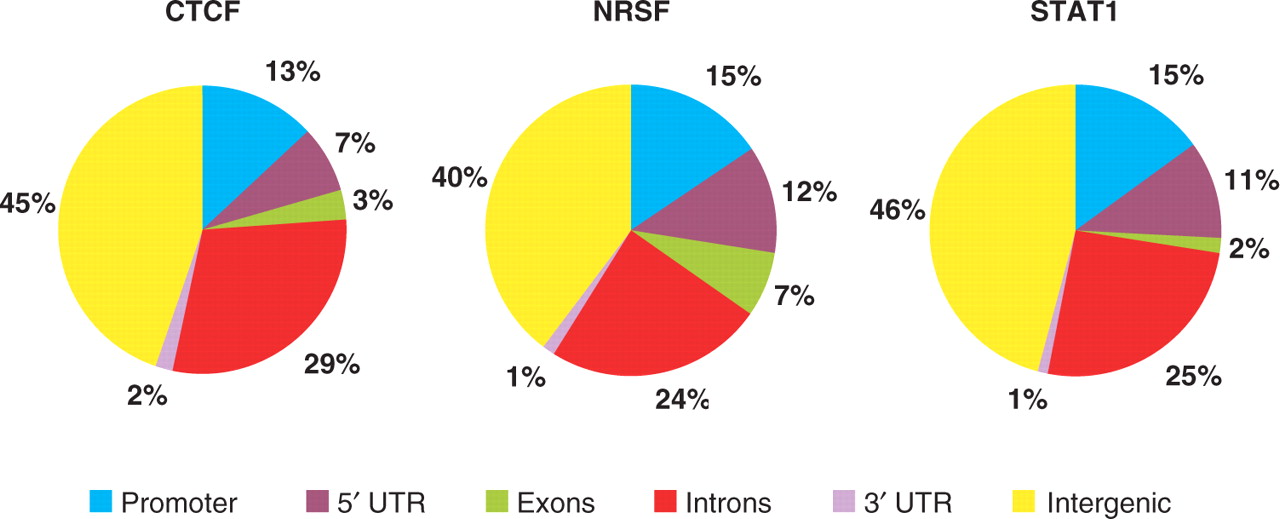
Jothi 2008, http://www.ncbi.nlm.nih.gov/pubmed/18684996
* Популярный R-пакет для геномной аннотации пиков: ChIPseeker
“Although the function of these regions has not been assessed, our work in Drosophila suggests that many are active enhancers, which trigger gene transcription. However, because factors can bind DNA without functional consequences, especially at HOT regions, the contribution of each of the bound factors to enhancer activity remains unclear.”
Translation: Not every piece of chewing gum attached to the soles of your shoes is functional. Moreover, the function of the sole of your shoe to which the chewing gum stuck is NOT to bind chewing gum.
Dan Graur (Judge Starling) commenting ENCODE 2014 paper.
Пример из жизни
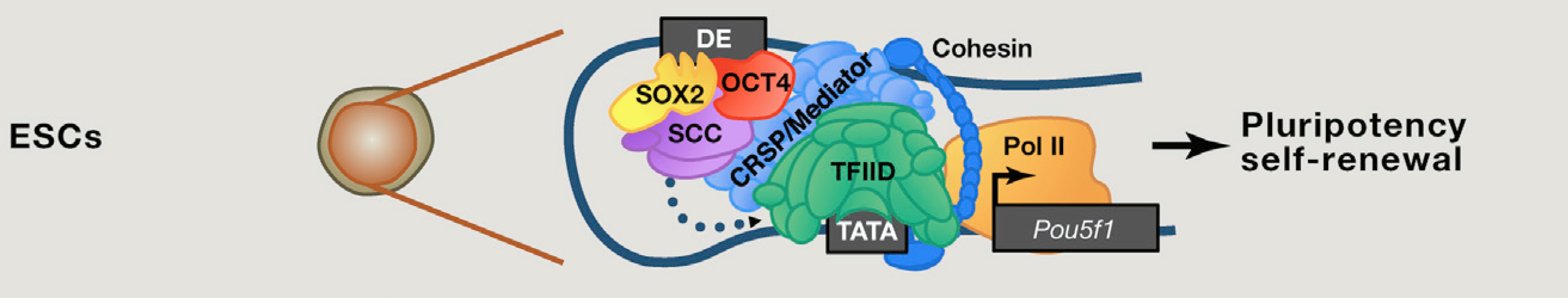

Пример из другой жизни
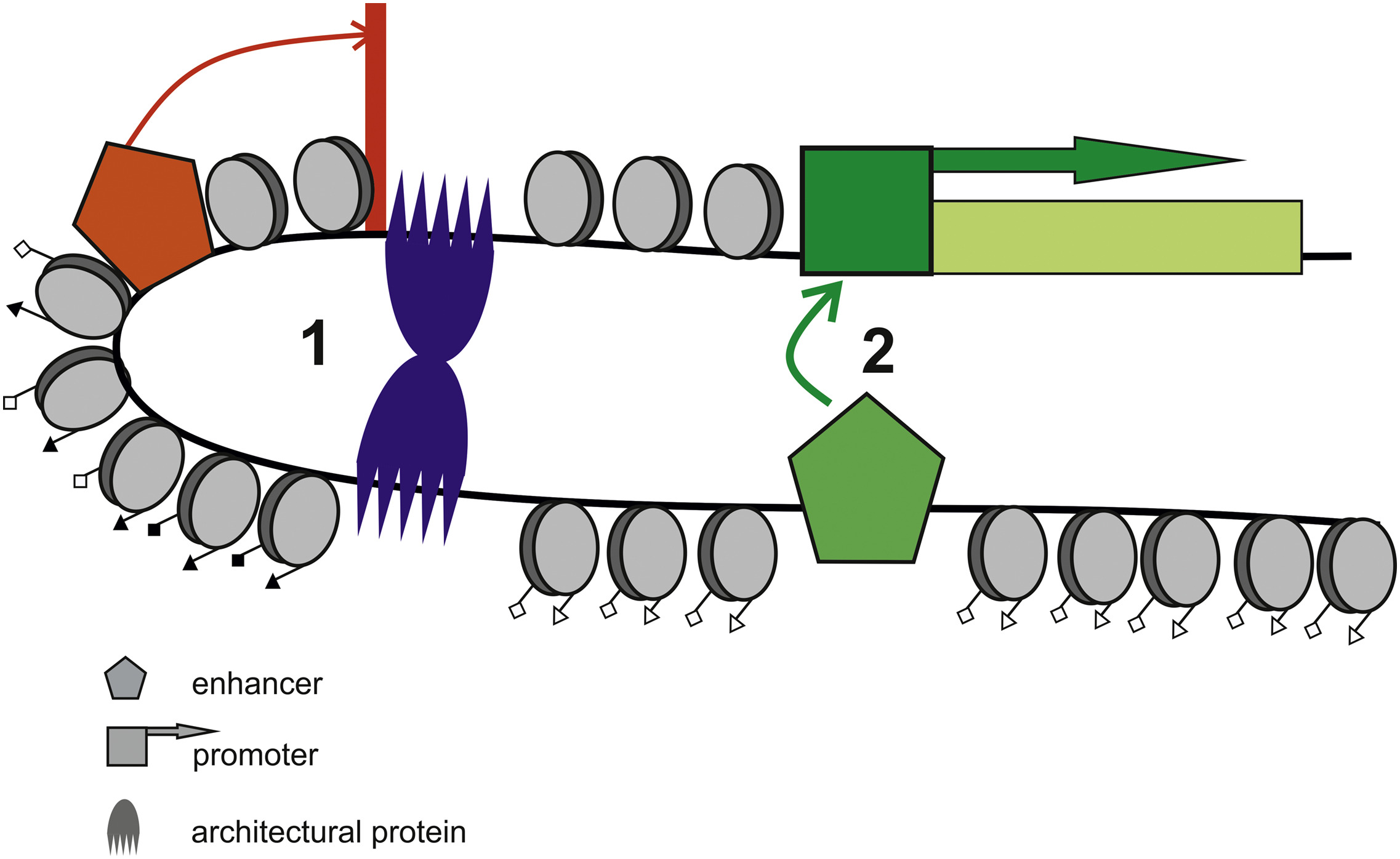
Maksimenko et al., BBA Gene Regulatory Mechanisms, 2020
Пример из другой жизни
Maksimenko et al., BBA Gene Regulatory Mechanisms, 2020
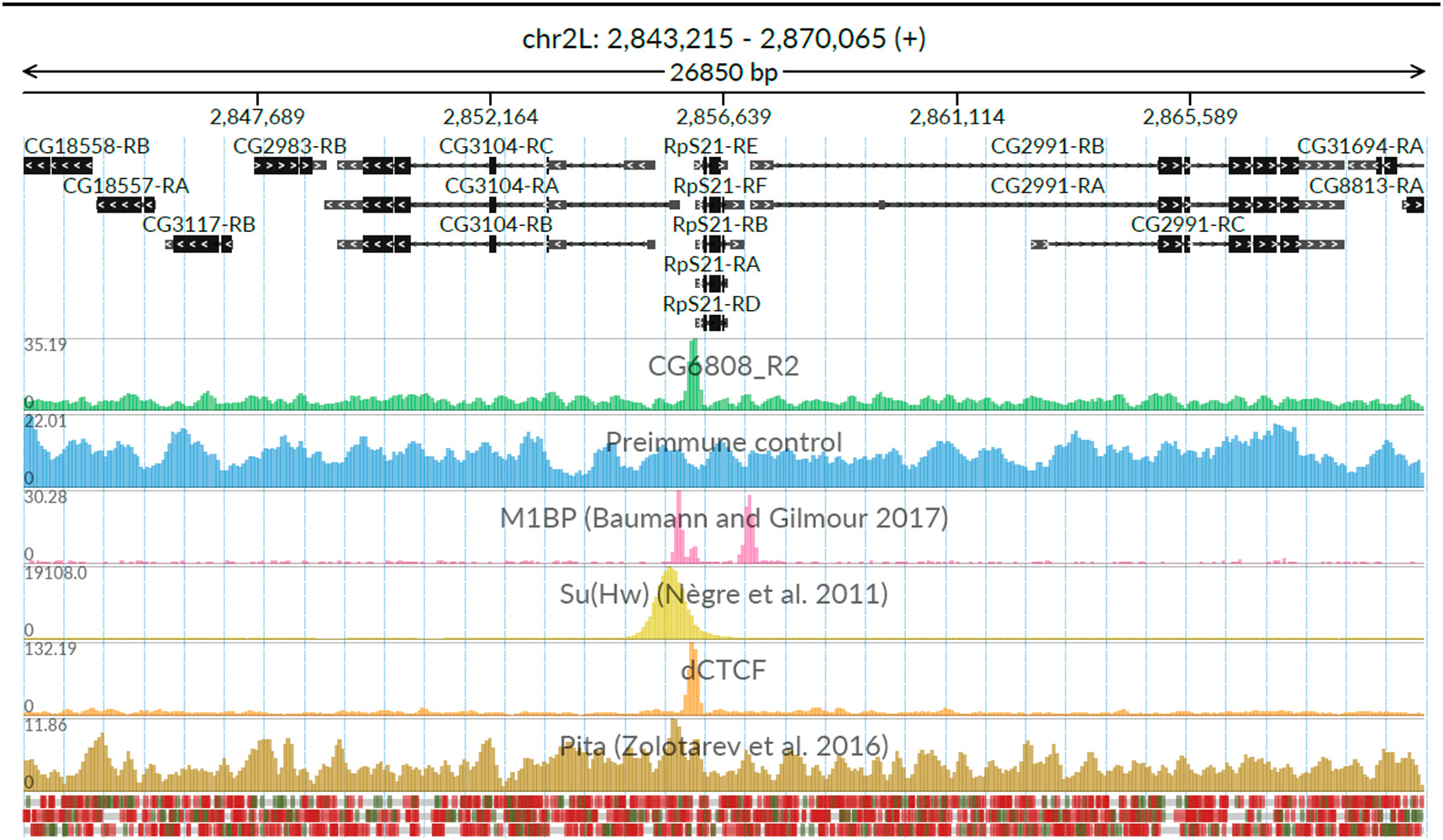
И красивые слайды
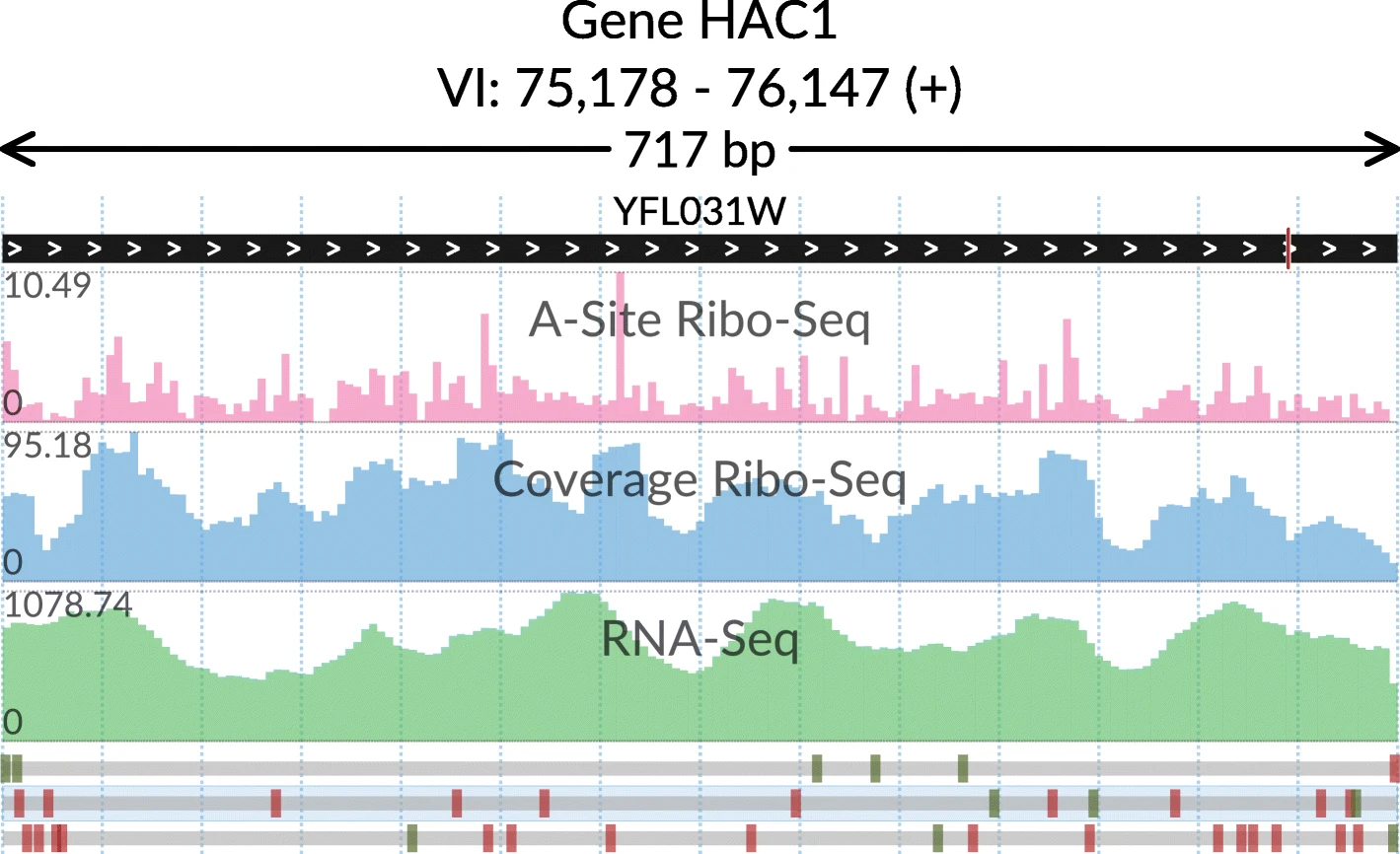
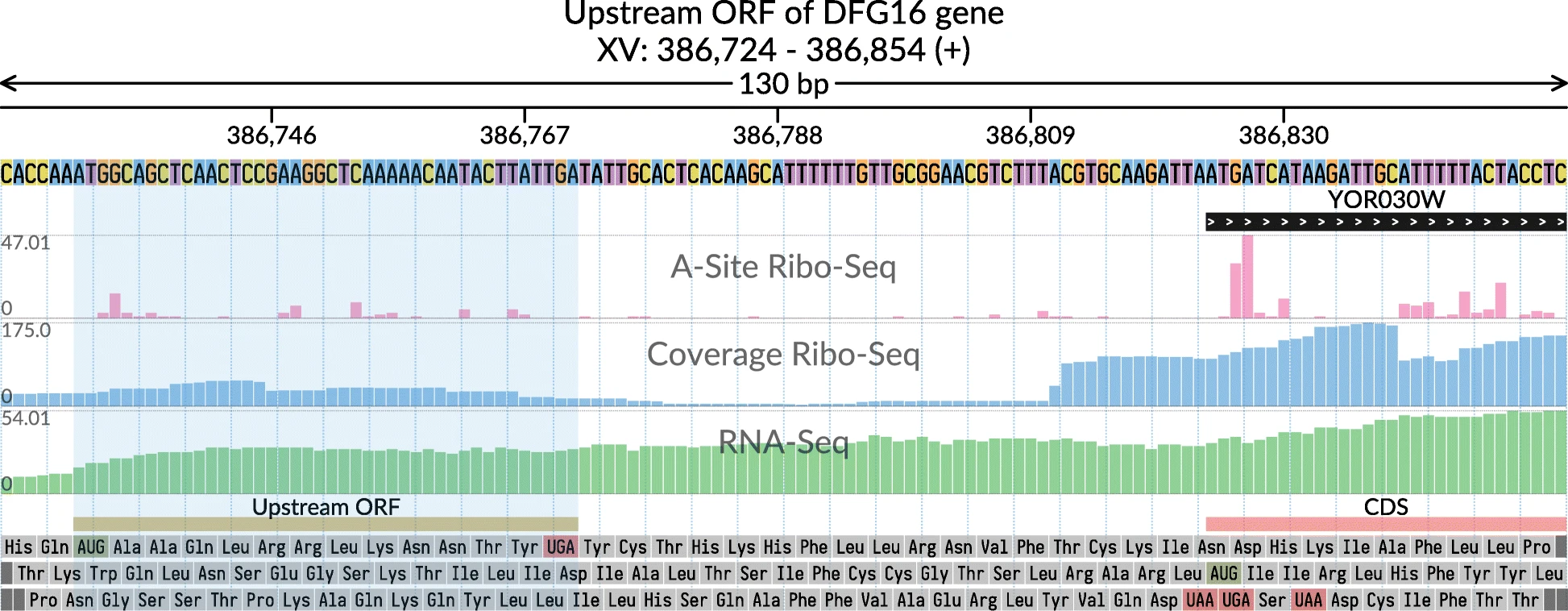
Egorov et al., BMC Bioinformatics, 2019
Egorov et al., BMC Bioinformatics, 2019
Самые полезные базы данных "про чипсеки"
ReMap - человек, арабидопсис (http://remap.univ-amu.fr/)
Cistrome - человек, мышь (http://cistrome.org/)
GTRD - все вподряд (http://gtrd.biouml.org/)
Каждый 1000й нуклеотид - отличается между двумя людьми (3 млн. замен). Но как сильно отличается регуляция генов?
Chen et al., Cell, 2012, doi:10.1016/j.cell.2012.02.009
Kasowski et al. 2010, Science
*Используя лимфобластоидные клеточные линии, получаемые при заражении В-лимфоцитов вирусом Эпштейна-Барр.
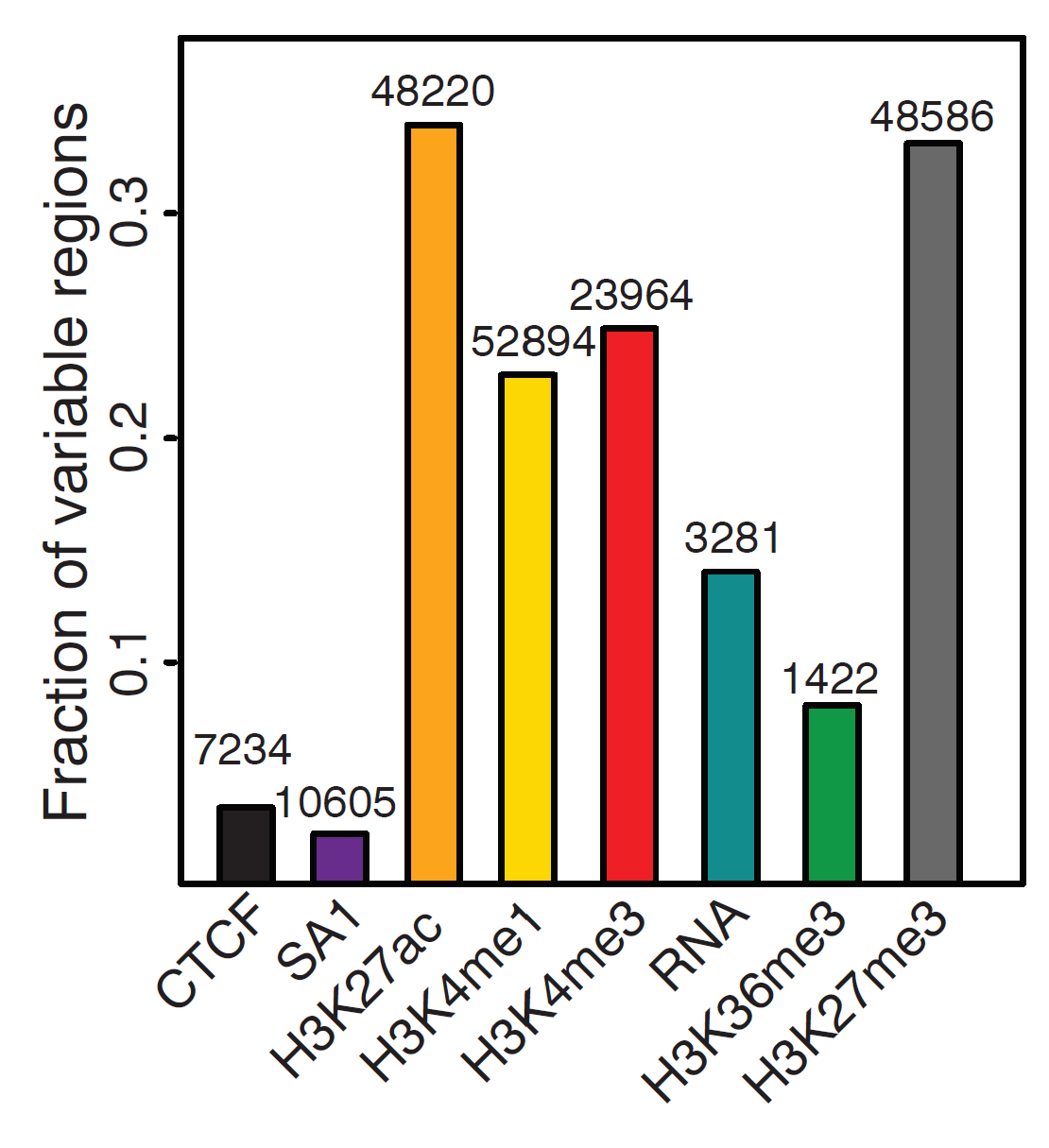
Kasowski et al. 2013, Science
Связывание факторов транскрипции определяется последовательностью!
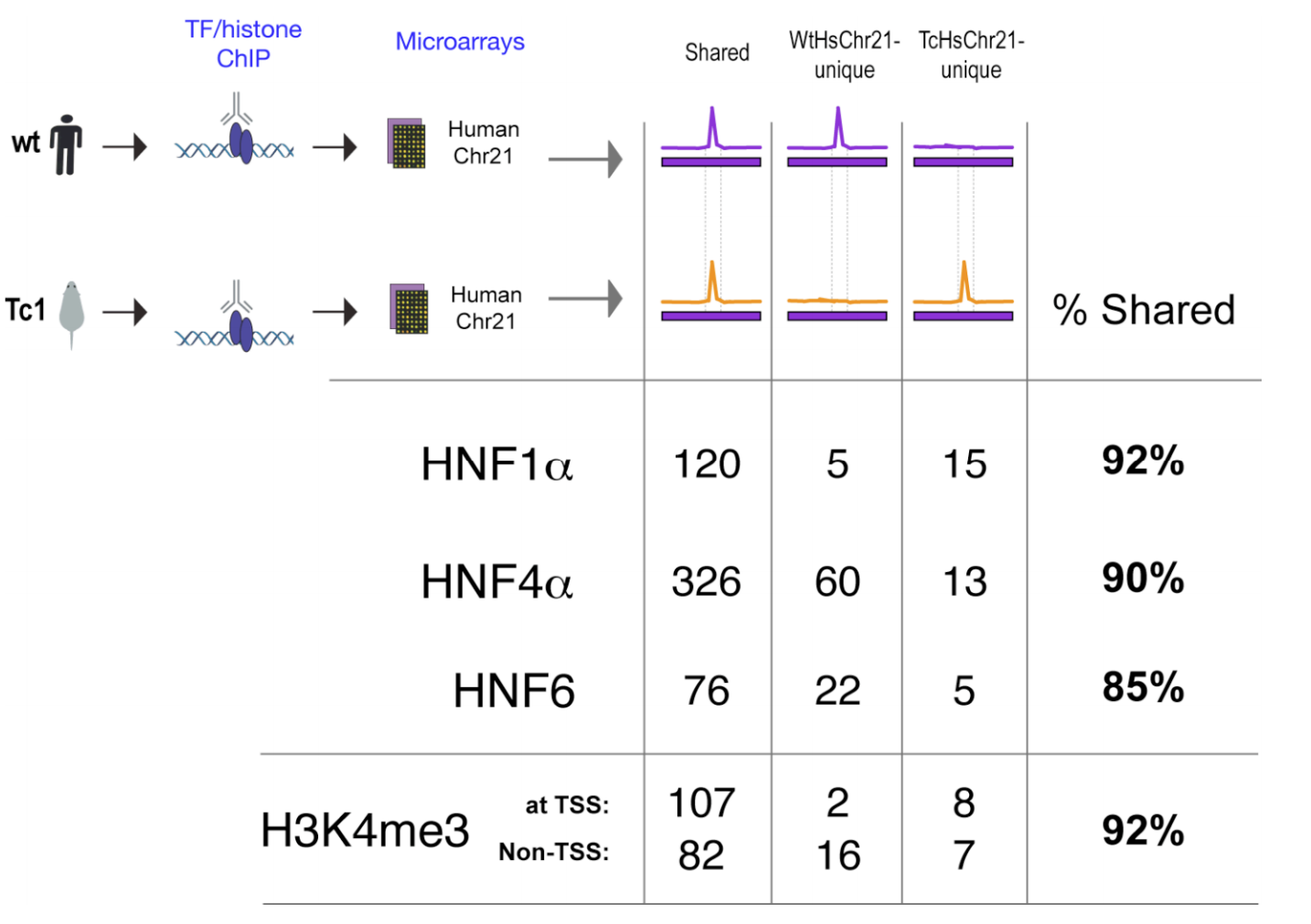
Wilson et al. 2018, Science
Take-home

Чипсеков уже сделано много и будет еще больше
Более продвинутые методы активно развиваются и "скоро все будет"
Интерпретация результатов чипсеков зависит от метода обработки данных и контролей
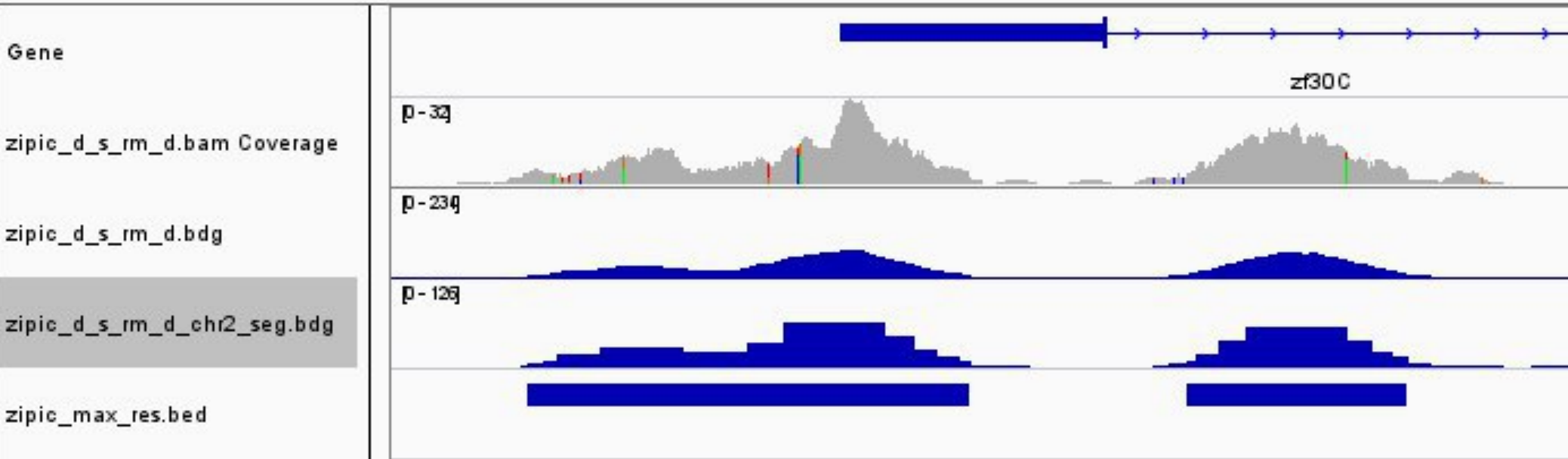
Распределение Пуассона - огого какое полезное (MACS, HOMER, pasio)
Анализ последовательностей - полезный независимый контроль результатов (3я серия)
Чипсеки - не только геномный адрес связывания белков (4я серия)